FAQs for mfold computations
- What does "initial dG" mean? What is the meaning of "dG = -yy.y [initially -xx.x]?
M. Zuker replies: For RNA folding, versions 3.x of mfold use free energy rules as described in:
D.H. Mathews, J. Sabina, M. Zuker & D.H. Turner
Expanded Sequence Dependence of Thermodynamic Parameters Improves Prediction of RNA Secondary Structure
J. Mol. Biol. 288, 911-940 (1999)
These rules are for folding at 37°C and are the default parameters that are used by the mfold web server.The rules for multi-branch loops are too complicated to be used in an efficient algorithm. Therefore, the folding is done using slightly simpler rules. This gives the "initial free energies" (initial dG). The free energies are then re-evaluated using the best rules that we have. They are reported as "dG = yy.y [initially -xx.x]", where 'yy.y' is the revised free energy, and 'xx.x' is the 'initial dG' found by the folding algorithm. In particular, the revised energies are determined by:
- using Jacobson-Stockmeyer theory to assign free energies to multi-branch loops that includes a term that grows logarithmically with the number of unpaired bases in the loop,
- and by computing coaxial stacking of adjacent helices in multi-branch loops or in the "exterior" loop. This latter feature has been implemented by David Mathews in his RNAstructure and related software.
On the mfold web server, the structures are ordered by their initial free energies. Another file, named "sort.ct", contains the same structures, but re-ordered according to the revised energies. You should consider the revised energies as "more correct", but when different structures are computed that are close in energy, there is no way to be confident that one is better than another unless additional methods are used, including experimental and computational.
As of February 10, 2006, the sorted structures are explicitly available through a hyperlink on the folding results page.
I noticed that some wobble base pairs do not form even if they are in the middle of a stem. It seems to happen with consecutive wobbles (GU followed by UG). Is it known to be impossible to form, or less favorable?
M. Zuker replies: For RNA folding, versions 3.x of mfold use free energy rules as described above. There are special free energy tables for 1 × 1 and 2 × 2 internal loops. Whenever you see a single, or two tandem potential wobble pairs that do not form, it means only that the free energy of the motif is determined from a special table and not from the usual base pair/base stacking free energy table. You should regard all "non-paired" GU or GU pairs in 1 × 1 or 2 × 2 interior loops as wobble pairs. Similarly, mismatched bases in such loops should be regarded as non-canonical base pairs.
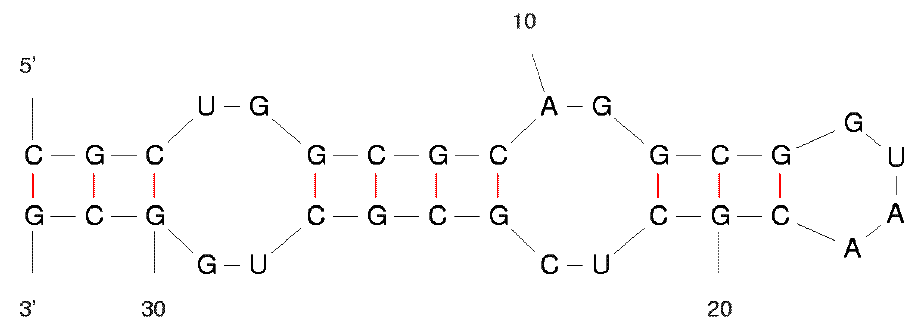
The two images on the left are equivalent. That it, there are two adjacent wobble base pairs and there is a non-canonical base pair next to a wobble pair. 
I have been trying to fold a microRNA sequence, but the server keeps returning an error
No folding is possible for 11Mar03-08-03-28
Job aborted! No Structure PlotsThe sequence is AACCACACAACCTACTACCTCA
I'm unsure if is due to the fact that there are no G's in the sequence or some unknown bug in the software.M. Zuker replies: My first comment is that miRNAs hybridize to a target, so I would not expect to find any secondary structure in a miRNA.
In the case of your sequence, the reported result of "No folding is possible" is correct. It has nothing to do with the lack of G's, although that is probably a contributing factor. The mfold and the newer UNAFold software filter out isolated base pairs. A base pair is isolated if no adjacent base pair is possible. This is the case with the sequence that you submitted. With the default filter option, no secondary structure is possible. If the filter is turned off, a folding containing from 1 to 3 isolated base pairs is possible. All of them are highly unstable and would never form. See also the comment about isolated base pairs with respect to folding with constraints.
I am trying to analyze multiple sequences at once, instead of having to analyze one at a time. Is there a way to do this? If so, how would I go about analyzing them all at once?
M. Zuker replies: You don't say what you wish to "analyze". That is "What computation(s) do you wish to perform on multiple sequences?"
There are 3 applications that allow you to enter multiple sequences and 1 that allows you to enter multiple pairs of sequences. All of these applications give you less than what you would get folding a single sequence or hybridizing two sequences.Zipfold: enter multiple sequences
Output: minimum free energy of each sequenceQuikfold: enter multiple sequences
Output: minimum free energy and close to minimum free energy structures for each sequence.Two-state melting (folding) : enter multiple sequences
Output: minimum free energy and structure for each sequence together with a melting temperature based on a crude two state model. (This output is much less than what is predicted when you use the application(s) that predict melting profiles.)Two-state melting (hybridization): enter multiple pairs of sequences
Output: minimum free energy and hybridization for each sequence pair together with a melting temperature based on a crude two state model. (same "much less" comment as above)