Applications


Next: Discussion Up: Using Reliability Information to Previous: Introduction
Applications
We chose a range of colors that vary fairly smoothly from red through orange, yellow, green, cyan, blue, magenta, and finally black. These colors are used to represent bases or base pairs that are ``well-determined'' to ``poorly-determined'', respectively. The exact correspondence between color and various ``well-determinedness'' indices is given in Figure 1. These colors were chosen for their visual appeal. The red, very ``well-determined'' regions catch the eye. The range of colors gives a good visual difference between pairs of discriminant measures.
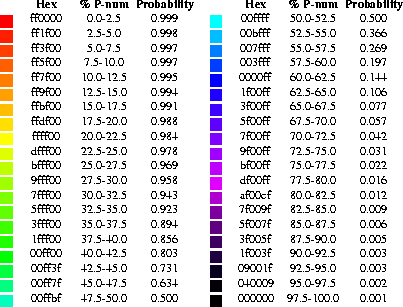
Figure 1: The color annotation that is
used to indicate the propensity of individual nucleotides to participate in base pairs
and whether or not a predicted base pair is well-determined. Forty colors that range
from red (unusually well-determined) to black (poorly determined) are used. Their
hexadecimal values are shown in column 1. The corresponding %P-num is shown in column
2. The %P-num value that is used to annotate each individual nucleotide of the
structure plot is calculated from the P-num table generated by the
mfold
package. Its absolute value depends on the energy range that the user selects in the
creation of the energy dot plot. The probability values shown in column 3 are used to
annotate structure with probability information from the Vienna RNA folding package. A
double logarithmic scale is used, as described in the text.
Initially we colored bases in structure plots. In this way, both the base identity and its level of ``well-determinedness'' could be shown simultaneously. Unfortunately, we found that the colors appeared too faintly in the annotated plots. In addition, the bases are too small to be seen in plots of large foldings. For this reason we chose to plot colored disks, or dots as we call them, that are roughly the size of the base characters they replace. The resulting plots are visually appealing and informative, even for very large molecules. Base coloring has nevertheless been retained as an option in the annotation programs.
When annotation is based on P-num or S-num, each base is colored according to its P-num or S-num value. We call this base dependent annotation. The P-num or S-num values are scaled linearly from 0 to 1 by dividing by the maximum. The colors are chosen according to a linear scale (Figure 1). In base dependent annotation methods, paired bases are not necessarily the same color. In a particular folding, one partner of a base pair might pair with only a few other bases in all close to optimal foldings, while the other partner might pair with many other bases. There is no a priori reason to expect symmetry.
Structure plots of two molecules of RNase P RNA [Reed et al., 1982,LaGrandeur et al., 1993,Brown, 1998] that were annotated with P-num values can be seen in Figures 2A and 2B. The annotated plot for E. coli (2A) shows that this structure is relatively poorly determined. Most dots are colored from light green to blue and some are dark purple. A few dots at the apex of several hairpins are bright red indicating that nucleotides within these small local regions are well-determined. In contrast, large helices in the annotated plot of the Sulfolobus acidocaldarius RNase P RNA (2B) are very well determined and are colored bright red. Additional features throughout the structure are colored orange and yellow indicating that they too are relatively well determined.
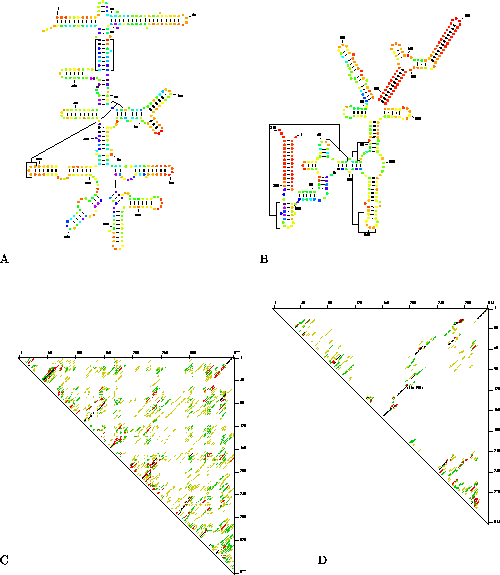
Figure 2: An illustration of how
well-determined the prediction is for two different molecules of RNase P RNA. A) A
structure plot for RNase P RNA from Escherichia coli annotated with P-num. B) A
structure plot for RNase P RNA from Sulfolobus acidocaldarius annotated with
P-num. C) The corresponding energy dot plot for the annotated structure plot shown in
2A. D) The energy dot plot corresponding to Figure 2B. The RNA structures shown in A and B are sub-optimal foldings.
They were selected from a group of automatically generated foldings based on their
consistency with the phylogenetic models. Thick lines were used to indicate base pairs
in these structures that correspond with base pairs in the published phylogenetic
models [Reed et al., 1982,LaGrandeur et al., 1993,Brown, 1998].
In Figures 2C and 2D we show the energy dot plots that correspond to the color annotations given in Figures 2A and 2B. The energy dot plots give the superposition of all base pairs within 5% of the optimal folding.They contain more information than the annotated structure plots (see Discussion), but are also more difficult to interpret. In each energy dot plot, the optimal folding is represented in black. Suboptimal base pairs are represented in color. Well determined features can be recognized easily because they are located in clear areas of the plot where few alternative base pairs form. Inspection of the dot plot for E. coli RNase P RNA (Figure 2C) shows that it is poorly determined; a uniform distribution of base pairs is seen at all levels of suboptimality. In contrast, the dot plot for Sulfolobus acidocaldarius RNase P RNA (Figure 2D) is relatively well determined. One small domain extending from nucleotides 116 to 193 (located in the center of plot near the diagonal) has virtually no competing base pairs. Similarly a long helix that pairs the 5' and 3' ends of the molecule is also well determined. These regions correspond to the helices shown in red in Figure 2B. In our own studies we use both the energy dot plot and the annotated structure plot of each folding prediction to analyze the folding potential of the predicted structure. The energy dot plot gives a good overview of the folding potential of the entire molecule, and the structure plot is used to extract detailed information about specific base paired regions.
Structure comparison annotation is achieved through a program called ss-compare. With it, base pairs in a reference structure are represented by thick lines that stand out from the non-conserved base pairs. The annotation can be used to illustrate differences between pairs of alternative predicted foldings, between wild-type and mutant genomes [Jacobson et al., 1998] and between phylogenetically determined versus computer predicted models of an RNA secondary structure. An example of the latter is shown in Figures 2A and 2B. In addition to color annotation, the structures have also been annotated with ss-compare to show whether any of the predicted helices are present in the published phylogenetic models for the two RNAs. In the E. coli plot 10 out of 16 of the helices that are shown in the plot are also found in the phylogenetic model for this RNA. In the S. acidocaldarius plot 8 helices out of a total of 12 helices are in the phylogenetic model. The structures that are shown are not optimal. They are selected from a group of automatically generated foldings. The selection criterion is the greatest degree of consistency with the published phylogenetic models for these RNAs. Approximately 60% of the predicted helices in the optimal foldings of the two RNAs match the phylogenetic models.
The program ss-compare can also be used to annotate a phylogenetic model of an RNA in order to examine how well individual structures are predicted. This feature is illustrated in Figure 3A where the phylogenetic model 16S ribosomal RNA from Thermus thermophilus [Murzina et al., 1988,Gutell, 1994] has been annotated with color to show how well determined individual base pairs are, and with thick lines to indicate which structural features are predicted correctly by mfold. The corresponding energy dot plot for this RNA is shown in Figure 3B. It is presented as an overlaid dot plot [Zuker & Jacobson, 1995] where predicted helices in the optimal folding are underscored in green if they are also found in the phylogenetic model. Large red lines indicate the position of helices in the phylogenetic model that are absent from the predicted optimal folding. It can be seen that suboptimal helices are located at many of these sites. Both the annotated structure plots (Figure 3A) and the energy dot plot (Figure 3B) of 16S rRNA from Thermus thermophilus show that the majority of local hairpins along the outer edges of the structure are extremely well determined and that all of these structures are predicted correctly. Poorly determined regions are located predominantly within the interior of the molecule, and only some of these helices were predicted correctly by mfold.
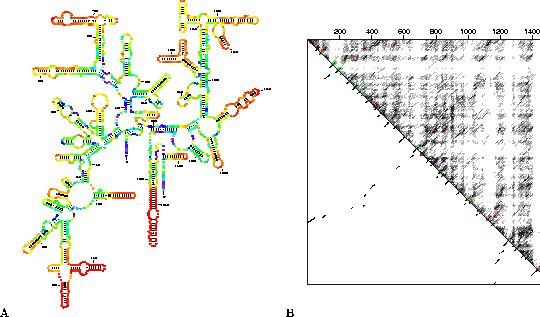
Figure 3: Annotation of the
phylogenetic model of 16S rRNA from Thermus thermophilus. A) Color annotation
based on P-num. The annotation is based on a 12 kcal energy dot plot created with
version 2.3 of mfold. Thick lines are used to show base pairs in the predicted
optimal folding of the RNA (not shown) that are also present in the phylogenetic model
[Murzina et al., 1988,Gutell, 1994]. B) A 12 kcal overlay energy dot plot. The
optimal predicted folding is shown in the lower left triangle. All base pairs within 12
kcal of the optimal folding are shown in the upper right triangle. Red and green lines
indicate the position of helices in the phylogenetic model.
Annotation can also be base pair dependent. This is especially
relevant when annotation is based on boxplot probabilities from the Vienna package.
Both bases in a base pair are given a color that corresponds to the probability of that
base pair. Single stranded bases are annotated according to the probability that they
are single stranded. P-num values can also be used in a base pair dependent way
by using and average value, 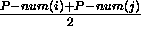 , to annotate
both bases, i and j, in a pair. When probabilities are used for
annotation, a double logarithmic scale is used to assign colors. For probabilities,
p, ranging from 0.999 to 0.5,
, to annotate
both bases, i and j, in a pair. When probabilities are used for
annotation, a double logarithmic scale is used to assign colors. For probabilities,
p, ranging from 0.999 to 0.5, 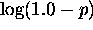 is
mapped linearly to the top 20 colors. For probabilities, p, in the range from
0.5 to 0.001,
is
mapped linearly to the top 20 colors. For probabilities, p, in the range from
0.5 to 0.001, 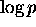 is mapped linearly to the
bottom 20 colors. This mapping makes it possible to distinguish easily among both high
and low probabilities.
is mapped linearly to the
bottom 20 colors. This mapping makes it possible to distinguish easily among both high
and low probabilities.
A base pair dependent annotation of the phylogenetic folding of 16S rRNA for Thermus thermophilus can be seen in Figure 4. Base pair probabilities were calculated with the Vienna RNA folding package using version 1.2.1. The corresponding color annotation is given in Figure 1. Overall, the plots shown in Figure 3A and in Figure 4 bear a striking resemblance to one another. Well determined hairpins at the edges of the structure are similar in position in both plots. A striking difference is observed, however, within the interior of the two plots. Many long range helices that are only predicted to be poorly determined by mfold, are predicted to be completely improbable (shown in black) with the Vienna package. The significance of these observed differences in the prediction by the two algorithms remain to be explored. It is useful to bear in mind that although both software packages are using almost identical energy functions, mfold computes what base pairs are possible in close to optimal foldings, while the Vienna package computes base pair probabilities.
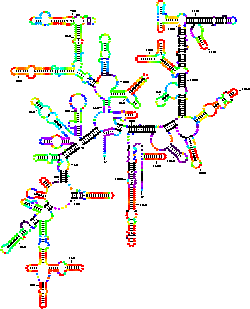
Figure 4: Color annotation of the
phylogenetic model for 16S rRNA of Thermus thermophilus based on probability.
The probability for each base pair was calculated using version 1.2.1 of the Vienna RNA
folding package. This program version uses energy rules that are identical to those use
in version 2.3 of mfold. The predicted folding is almost indistinguishable from
the predicted optimal folding that is generated with version 2.3 of
mfold.
The final annotation method used is called helix dependent. All the bases in a helix are colored according to the H-num value of the helix. As with P-num and S-num, the numbers are divided by the largest value, and thus range between 0 and 1. No example is shown for this annotation.
Next: Discussion Up: Using Reliability Information to Previous: Introduction
 |
Michael Zuker Institute for Biomedical Computing Washington University in St. Louis August 21 1998. |